







Belite Bio, Inc announced topline results from the global phase 3 DRAGON trial of tinlarebant, marking the first successful pivotal trial in patients with Stargardt disease type 1 (STGD1), the company said in a press release.
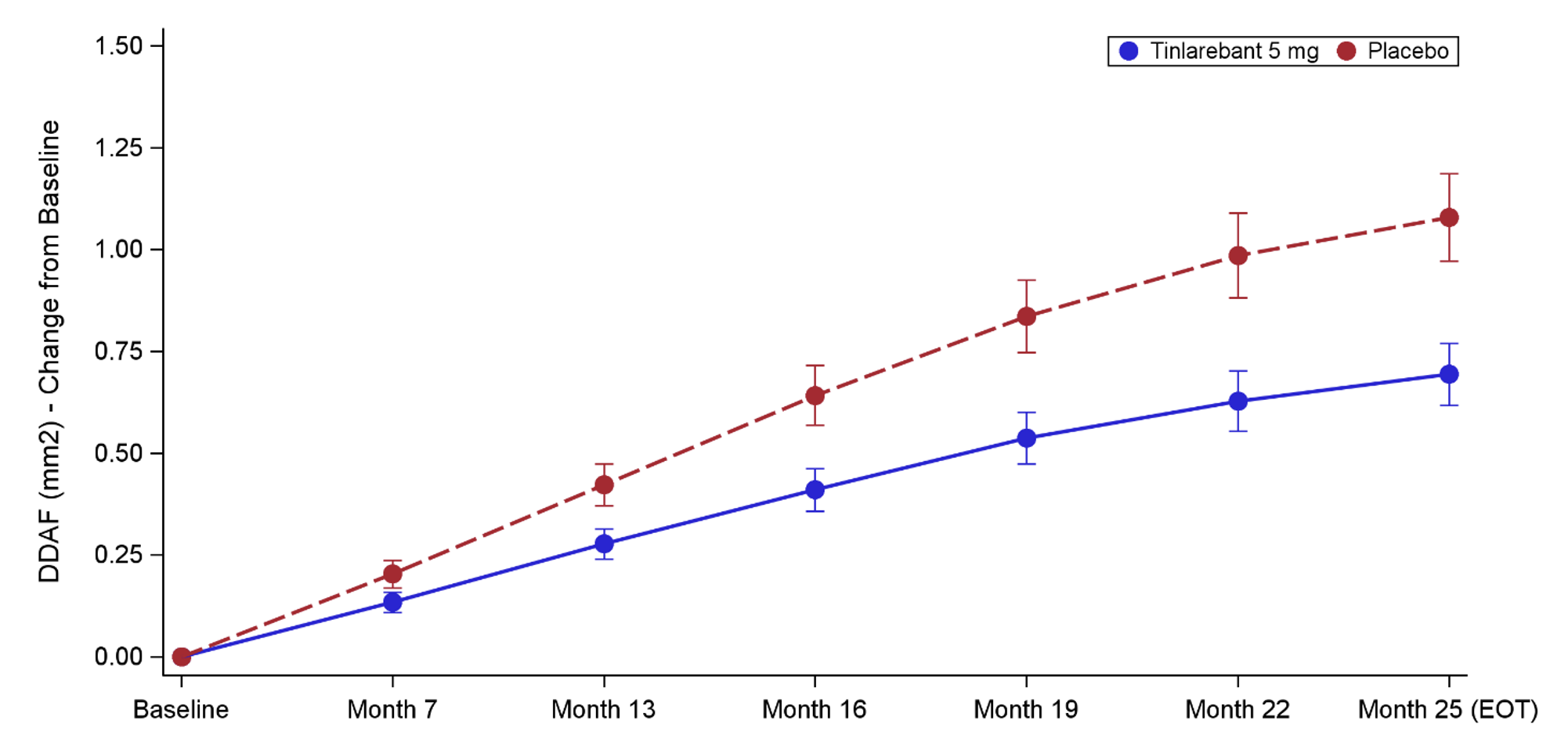
DRAGON enrolled 104 patients with STGD1 and met its primary efficacy endpoint, demonstrating a statistically significant and clinically meaningful 36% reduction in the growth rate of retinal lesions, measured as definitely decreased autofluorescence (DDAF) by fundus autofluorescence imaging, compared with placebo. Statistical significance was reached when applying the prespecified analysis (P=.0033). Considering the progressive nature typically seen in STGD1, a further post hoc analysis providing a specific data correlation showed that the treatment effect remained consistent (P<.0001), according to the company.
As expected, the company said, the overall change in visual acuity was minimal over the period of 24 months in both study groups, consistent with natural history data. The safety profile remains consistent with previous trials, and tinlarebant was well tolerated with 4 treatment-related discontinuations. After the full analysis is complete, the company said it plans to share additional data at upcoming medical meetings.
Belite Bio said it plans to engage regulatory authorities to discuss potential next steps and to submit new drug applications for tinlarebant in the first half of 2026. Tinlarebant has been granted Breakthrough Therapy, Fast Track, and Rare Pediatric Disease Designations in the United States; Orphan Drug Designation in the US, Europe, and Japan; and Pioneer Drug Designation in Japan for STGD1.
According to the company, the DRAGON trial was a 24-month, randomized (2:1, active: placebo), double-masked, placebo-controlled, global, multi-center, pivotal phase 3 trial in adolescent STGD1 patients.
Regarding patient demographics, 104 patients (n=69 in tinlarebant arm and n=35 in placebo arm), ranging in age from 12 to 20 years, were enrolled in DRAGON. Also, all patients had been diagnosed with STGD1 with at least 1 mutation identified in the ABCA4 gene, an atrophic lesion size within 3 disc areas (7.62 mm2), and a best-corrected visual acuity (BCVA) of 20/200 or better.
Regarding positive efficacy results, tinlarebant achieved the primary efficacy endpoint demonstrating a statistically significant reduction in lesion growth rate of 35.7% vs placebo (P=.0033) as measured by retinal imaging, when applying an unstructured covariance matrix under the Mixed Model for Repeated Measures (MMRM). To account for the longitudinal nature of the collected data while maintaining model stability given the sample size in the DRAGON trial, a post hoc analysis using an autoregressive covariance matrix under MMRM yielded a treatment effect size of 35.4% (P<.0001).
The company also said a statistically significant treatment effect was also observed in the fellow eye for the primary endpoint with 33.6% lesion growth reduction (P=.041). In addition, tinlarebant slowed decreased autofluorescence (DAF) lesion growth, the key secondary endpoint calculated as the sum of DDAF and questionably decreased autofluorescence (QDAF), in the study eye by 33.7% (P=.027) and in the fellow eye by 32.7% (P=.017).
The 5 mg daily dose achieved a reduction in RBP4 levels by a mean of approximately 80% relative to baseline. Also, retinal binding protein 4 (RPB4) levels returned to 84% of the baseline value at end of study (1 to 3 months following drug cessation). Recovery of RBP4 concentration correlated well with the decreased tinlarebant exposure.
According to the company, the strong safety profile was consistent with past trials. Tinlarebant (5 mg orally, daily) was well tolerated in adolescent STGD1 patients. There were no drug or trial discontinuations due to nonocular adverse events (AE). In addition, there were 4 drug discontinuations that were related to the treatment. Xanthopsia and delayed dark adaptation are the most common drug-related ocular AE. The majority of xanthopsia, delayed dark adaptation, and night vision impairment were mild, and most resolved during the trial. Headaches were the most commonly reported treatment-related non-ocular AE, the company said.